Biotecnología Industrial y Microbiología Aplicada
Biotecnología Industrial y Microbiología Aplicada
TRABAJO PRÁCTICO N°1
Parte D ALINEAMIENTO ESTRUCTURAL DE ENZIMAS
La investigación de los determinantes estructurales de la adaptación enzimática a la temperatura es un requisito fundamental tanto en términos de investigación aplicada como industrial para desarrollar nuevos biocatalizadores activos en diferentes rangos de temperatura. Un alineamiento estructural es un tipo de alineamiento de secuencias basado en la comparación de la forma. Estos alineamientos intentan establecer equivalencias entre dos o más estructuras de polímeros basándose en su forma y conformación tridimensional. El proceso se aplica normalmente a las estructuras terciarias de las proteínas
Es una valiosa herramienta para la comparación de proteínas con baja similitud entre sus secuencias, en donde las relaciones evolutivas entre proteínas no pueden ser fácilmente detectadas por técnicas estándares de alineamiento de secuencias.
Los alineamientos estructurales pueden comparar dos o múltiples secuencias. Puesto que estos alineamientos dependen de información sobre todas las conformaciones tridimensionales de las secuencias problema, el método sólo puede ser usado sobre secuencias donde estas estructuras sean conocidas. Estas se encuentran normalmente por cristalografía de rayos X o espectroscopia de resonancia magnética nuclear. Si bien es posible realizar un alineamiento estructural sobre estructuras producidas mediante métodos de predicción de estructura
Los alineamientos estructurales son especialmente útiles para analizar datos surgidos de los campos de la genómica estructural y de la proteómica, y pueden usarse como puntos de comparación para evaluar alineamientos generados por métodos bioinformáticos basados exclusivamente en secuencias.
OBJETIVO: Estudio de alineamiento estructural comparativo de subtilisin-like serine-proteasas procedentes de microrganismos psicrófilos, mesófilos y termófilos,
basado en una publicación científica (Tiberti & Papaleo, Journal of Structural Biology 174 (2011) 69–83), emplea herramientas virtuales de acceso libre en la web.
Responda las preguntas en rojo en cada punto.
Parte D1: Obtención de las estructuras 3D de las proteínas a ser evaluadas y alineamiento estructural de a pares
Desde RCSB PDB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do)
Es posible realizar búsquedas simples y avanzadas basadas en las anotaciones correspondientes a la secuencia,
estructura y función, y para visualizar, descargar y analizar moléculas.
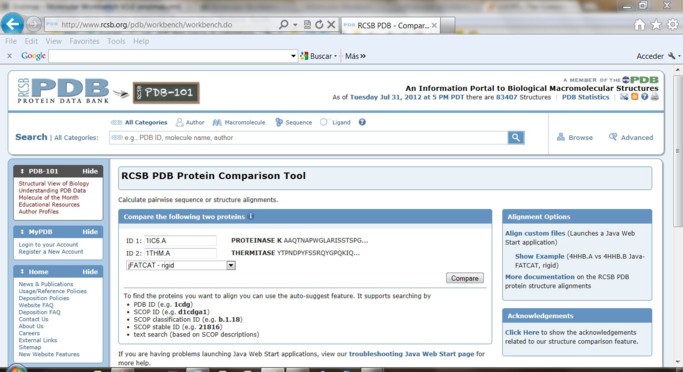
Metodología 1) Ingrese en RCSB PDB 2) En el campo de búsqueda ingrese el PDB ID de la proteína buscada. Empiece con 1IC6. Desde la solapa "Sequence" puede visualizar la estructura 3D con el Jmol con el botónen modo similar al que se muestra. Con el botón derecho del mouse puede modificar la visualización
El sitio permite visualizar los sitios de unión a ligandos y sus cadenas laterales de interacción.
Desplegando el menu "Add Annotations" puede incorporar más opciones de visualización con distintos métodos de predicción
En nuestro caso evaluaremos 3 proteínas: 1SH7, 1THM, 1IC6. Habrá para ello 3 pestañas simultaneas en el navegador.
3) Tome nota de las características de cada una de las 3 proteínas.
Describa las tres proteínas. Identifíquelas y complete los datos solicitados en una tabla.
Característica Térmica, % Hélices, % Hoja Beta
Se describe el sitio catalítico en las tres proteínas?
4) Desde el menú desplegable Download Files ubicado junto al PDB ID seleccione Fasta sequence y guárdelo. Luego PDB File (Text). Guarde como sólo texto el archivo y modifique la extensión a .pdb. Realice lo mismo para las 3 proteínas.
Los archivos en formato Fasta serán requeridos más adelante en el TP.
Conservar los archivos .pdb le permite desde su maquina analizar alineamientos de estructura 3D de las proteínas
Puede emplear algún programa libre disponible en la web si bien no se emplearán en el TP:
Chimera (http://www.cgl.ucsf.edu/chimera/) o VMD (http://www.ks.uiuc.edu/Research/vmd/)
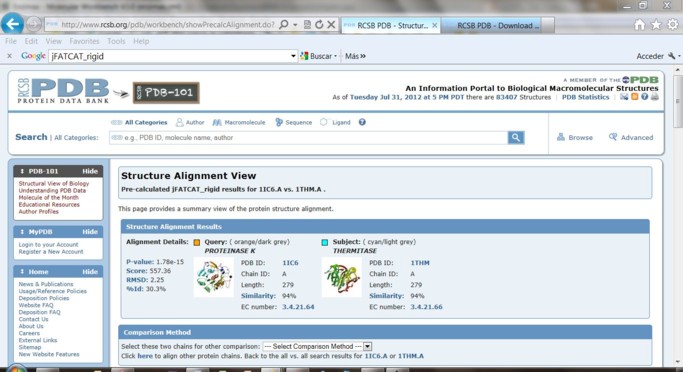
5) Alineamiento de las estructuras 3D de las proteínas empleando Protein Comparison Tool. Coloque el nombre de PBD ID de la proteína mesófilas y compárela con las dos extremófilos. Para esto seleccione el método jFATCAT_rigid y realice la comparación de a pares apretando la tecla
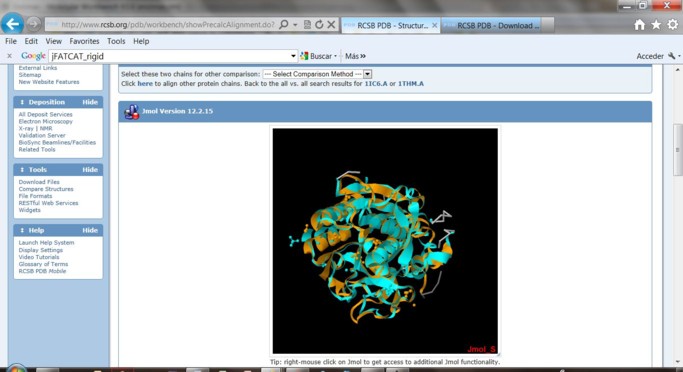
6) En la pantalla aparecerá la visualización con el Jmol. Este plugin depende de que el entorno Java se encuentre instalado en su computadora.
Para ello verifique los requerimientos en Browser compatibility Check
En la visualización obtenida cada una de las estructuras y sus ligandos están presentados en un color diferente.
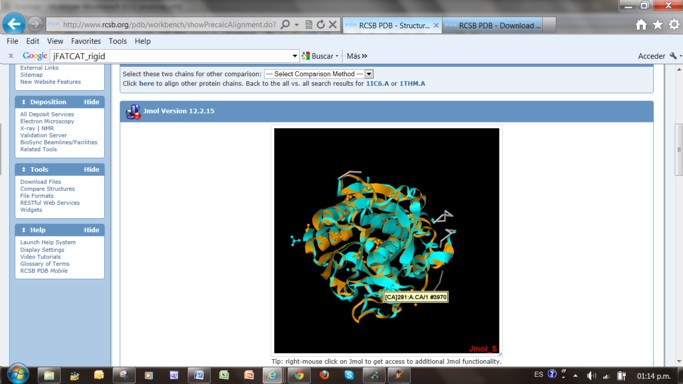
Identifique los átomos de Ca2+ ligandos en cada uno de los pares de proteínas con el cursor.
Puede hacerlo desde el menú con el mouse derecho identificando los elementos en la visualización
Entre en "Elementos" y buscar todos los ligandos, luego en "Seleccionar", "ver solo seleccionados"
6) Repita el procedimiento para el par restante
Indique los ligandos de cada estructura 3D
Puede utilizar el programa en línea desde el link para hacerlo en su computadora, Structural Aligment opción Align custom files
Parte D2: Alineamiento de las estructuras 3D de las proteínas empleando DALI. Un método de alineamiento estructural común y popular es DALI (de Distance ALIgnment matrix, o matriz de alineamiento de distancias),
que rompe las estructuras problema en fragmentos de hexapéptidos y calcula una matriz de distancia evaluando los patrones de contacto
entre fragmentos sucesivos.
Cuando las matrices de distancia de dos proteínas comparten las mismas o similares características en aproximadamente las mismas posiciones,
puede decirse que tienen similares plegamientos con bucles de longitud similar conectando sus elementos de estructura secundaria. Es posible obtener familias de proteínas estructuralmente similares, en la que todas las estructuras de proteínas conocidas son alineadas unas con
otras para determinar sus vecinas estructurales y la clasificación de los plegamientos. Hay una base de datos utilizable basada en DALI que realiza el alineamiento estructural “todos contra todos” los registros de estructuras 3D en la
base de datos PDB generando como resultado los vecinos estructurales (http://ekhidna.biocenter.helsinki.fi/dali/start). Es posible también emplear la versión autónoma conocida como DaliLite para alineamientos de a pares
(http://ekhidna.biocenter.helsinki.fi/dali_lite/start).
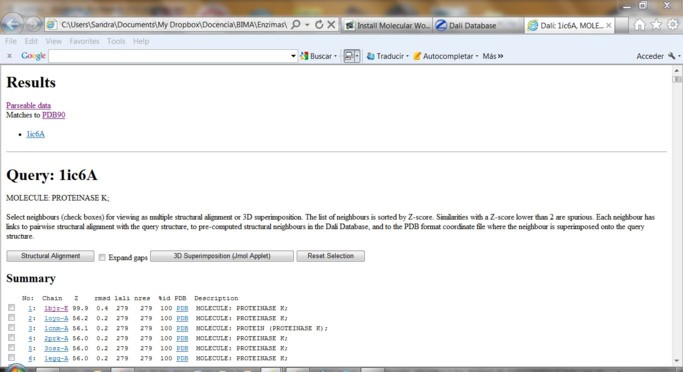
Metodología 1) Ingrese en DALI Database 2) Utilice uno de los PDB ID del paso anterior como criterio de búsqueda
3) El resultado le provee un listado de vecinos estructurales con distinto grado de identidad.
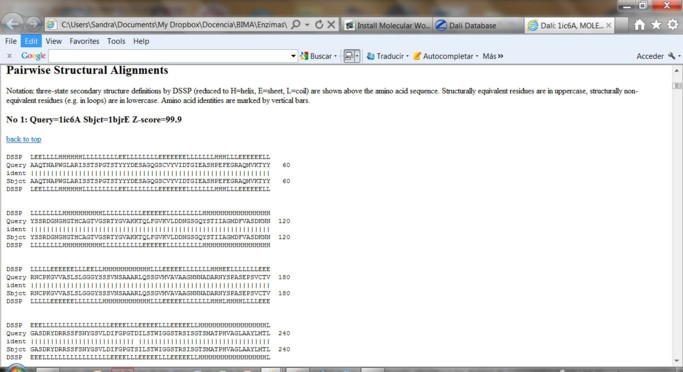
Al final del listado se presentan los alineamientos estructurales de a pares en base a la estructura secundaria donde H=helix, E=Beta-sheet y L=coil.
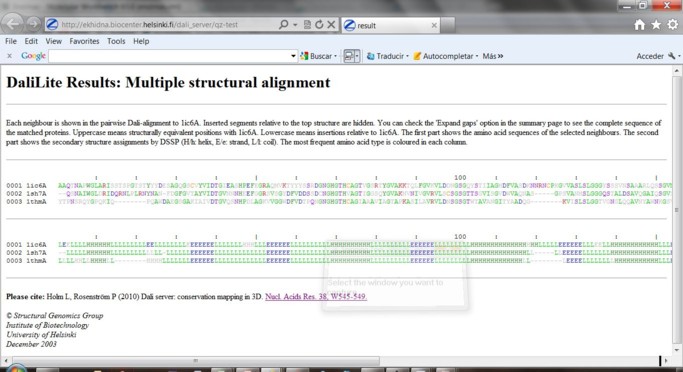
4) Seleccione los casilleros de las otras 2 proteínas. Utilizando el Botónpermite observar el alineamiento estructural múltiple.
Realice el alineamiento de la secuencia primaria de las tres proteínas con el programa ClustalO (http://www.ebi.ac.uk/Tools/msa/clustalo/).
Para ello debe construir un archivo en formato FASTA de múltiples secuencias con los archivos Fasta que obtuvo del RCSB PDB.
Analice ese alineamiento de la secuencia primaria y compárelo con el resultado de DALI en estructura secundaria.
Qué características observa en cuanto a la composición de estructuras secundarias entre las 3 proteínas?
Se corresponden con las secuencias primarias que se conservan?
Utilice el botónpara visualizar el alineamiento múltiple con el programa Jmol Para la visualización el programa depende de la plataforma Java que debe estar instalada en la máquina
5) En la visualización obtenida cada una de las estructuras y sus ligandos están presentados en un color diferente. Identifique los átomos de Ca2+ unidos a cada una de las tres proteínas.
Existe superposición de los atómos de Ca2+ entre los distintos pares?
6) Presentar un informe en formato digital por grupo de trabajo con las respuestas y conclusiones obtenidas a sandra@qb.fcen.uba.ar
Referencias
Feller, J. Phys.: Condens. Matter 22 (2010) 323101 (17pp)
Tiberti & Papaleo, Journal of Structural Biology 174 (2011) 69–83
Ye & Godzik, Bioinformatics. (2003) Oct;19 Suppl 2:ii246-55.
Holm & Rosenström, Nucl. Acids Res. 38, (2010) W545-549